Содержание статьи:

Синдром Эдвардса (трисомия 18). Причины, симптомы, признаки, диагностика и лечение патологии
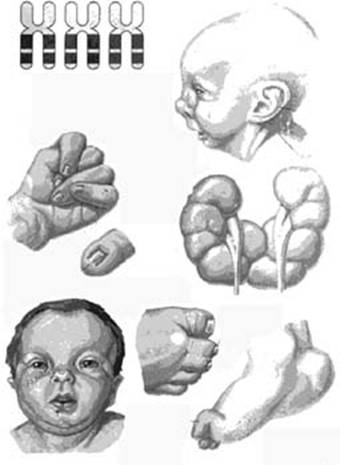 Синдром Эдвардса или трисомия 18 представляет собой тяжелое врожденное заболевание, вызванное хромосомными нарушениями. Оно является одной из наиболее распространенных патологий в данной категории (уступает по частоте лишь синдрому Дауна). Заболевание характеризуется многочисленными нарушениями в развитии различных органов и систем. Прогноз для ребенка обычно неблагоприятный, но многое зависит от ухода, который ему способны обеспечить родители.
Синдром Эдвардса или трисомия 18 представляет собой тяжелое врожденное заболевание, вызванное хромосомными нарушениями. Оно является одной из наиболее распространенных патологий в данной категории (уступает по частоте лишь синдрому Дауна). Заболевание характеризуется многочисленными нарушениями в развитии различных органов и систем. Прогноз для ребенка обычно неблагоприятный, но многое зависит от ухода, который ему способны обеспечить родители.Распространенность синдрома Эдвардса по земному шару варьирует от 0,015 до 0,02%. Четкой зависимости от местности или расы не наблюдается. Статистически девочки болеют в 3 – 4 раза чаще мальчиков. Научного объяснения этой пропорции пока не выявлено. Тем не менее, отмечен ряд факторов, которые могут повысить риск возникновения этой патологии.
Как и другие хромосомные мутации, синдром Эдвардса является, в принципе, неизлечимым заболеванием. Самые современные методы лечения и ухода могут лишь поддерживать жизнь ребенка и способствовать определенному прогрессу в его развитии. Единых рекомендаций по уходу за такими детьми нет из-за огромного разнообразия возможных нарушений и осложнений.
Интересные факты
- Описание основных симптомов данной болезни было сделано еще в начале XX века.
- До середины 1900-х годов собрать достаточную информацию об этой патологи не представлялось возможным. Во-первых, для этого необходим был соответствующий уровень развития технологий, который позволил бы обнаружить лишнюю хромосому. Во-вторых, большинство детей умирало в первые дни или недели жизни из-за низкого уровня оказания медицинской помощи.
- Первое полное описание болезни и ее основной причины (появление лишней 18-й хромосомы) было сделано только в 1960 году врачом Джоном Эдвардом, в честь которого тогда и назвали новую патологию.
- Реальная частота синдрома Эдвардса составляет 1 случай на 2,5 – 3 тысячи зачатий (0,03 – 0,04%), однако официальные данные значительно ниже. Это объясняется тем, что почти половина эмбрионов с данной аномалией не выживают и беременность заканчивается спонтанным абортом или внутриутробной смертью плода. Подробная диагностика причины выкидыша при этом проводится редко.
- Трисомия представляет собой вариант хромосомной мутации, при которой у человека в клетках содержится не 46, а 47 хромосом. Существует всего 3 синдрома в данной группе заболеваний. Помимо синдрома Эдвардса это синдромы Дауна (трисомия 21 хромосомы) и Патау (трисомия 13 хромосомы). При наличии других добавочных хромосом патология несовместима с жизнью. Лишь в этих трех случаях возможно рождение живого ребенка и его дальнейший (хоть и замедленный) рост и развитие.
Причины генетической патологии
 Синдром Эдвардса является генетическим заболеванием, которое характеризуется наличием дополнительной хромосомы в геноме человека. Чтобы разобраться в причинах, которые вызывают видимые проявления этой патологии, необходимо выяснить, что представляют собой собственно хромосомы и генетический материал в целом.
Синдром Эдвардса является генетическим заболеванием, которое характеризуется наличием дополнительной хромосомы в геноме человека. Чтобы разобраться в причинах, которые вызывают видимые проявления этой патологии, необходимо выяснить, что представляют собой собственно хромосомы и генетический материал в целом.В каждой клетке человека имеется ядро, которое отвечает за хранение и обработку генетической информации. В ядре содержится 46 хромосом (23 пары), которые представляют собой многократно упакованную молекулу ДНК (дезоксирибонуклеиновая кислота). Эта молекула содержит в себе определенные участки, называемые генами. Каждый ген является прототипом определенного белка в организме человека. При необходимости клетка считывает информацию с этого прототипа и вырабатывает соответствующий белок. Дефекты генов ведут к производству аномальных белков, которые и ответственны за появление генетических заболеваний.
Хромосомная пара состоит из двух идентичных молекул ДНК (одна отцовская, другая – материнская), которые сцеплены между собой небольшим мостиком (центромерой). Место сцепления двух хромосом в паре предопределяет форму всего соединения и его вид под микроскопом.
Все хромосомы хранят различную генетическую информацию (о разных белках) и подразделяются на следующие группы:
- группа А включает 1 – 3 пару хромосом, которые отличаются большими размерами и X-образной формой;
- группа В включает 4 – 5 пару хромосом, которые также являются крупными, но центромера лежит дальше от центра, из-за чего форма напоминает букву Х со смещенным вниз или вверх центром;
- группа С включает 6 – 12 пару хромосом, которые по форме напоминают хромосомы группы В, но уступают им по размерам;
- группа D включает 13 – 15 пару хромосом, для которых характерны средние размеры и расположение центромеры у самого конца молекул, что придает сходство с буквой V;
- группа Е включает 16 – 18 пару хромосом, которые характеризуются маленькими размерами и срединным расположением центромеры (форма буквы Х);
- группа F включает 19 – 20 хромосомные пары, которые несколько мельче хромосом группы Е и схожи с ними по форме;
- группа G включает 21 – 22 пару хромосом, которые характеризуются V-образной формой и очень маленькими размерами.
Синдром Эдвардса относится к так называемым хромосомным заболеваниям, когда проблема состоит не в дефекте гена, а в дефекте целой молекулы ДНК. Если быть более точным, то классическая форма этого заболевания подразумевает наличие лишней 18-й хромосомы. Кариотип в таких случаях обозначается как 47,ХХ, 18+ (для девочки) и 47,ХY, 18+ (для мальчика). Последняя цифра обозначает номер добавочной хромосомы. Излишек генетической информации в клетках приводит к появлению соответствующих проявлений болезни, которые и объединены под названием «синдром Эдвардса». Наличие дополнительной (третьей) хромосомы под номером 18 дало другое (более научное) название болезни – трисомия 18.
В зависимости от формы хромосомного дефекта различают три вида данного заболевания:
- Полная трисомия 18. Полная или классическая форма синдрома Эдвардса предполагает, что все клетки в организме имеют дополнительную хромосому. Данный вариант заболевания встречается более чем в 90% случаев и является наиболее тяжелым.
- Частичная трисомия 18. Частичная трисомия 18 является весьма редким феноменом (не более 3% от всех случаев синдрома Эдвардса). При ней в клетках организма содержится не целая дополнительная хромосома, а лишь ее фрагмент. Такой дефект может быть результатом неправильного деления генетического материала, но встречается он очень редко. Иногда часть восемнадцатой хромосомы присоединяется к другой молекуле ДНК (внедряется в ее структуру, удлиняя молекулу, или просто «цепляется» с помощью мостика). Последующее деление клеток приводит к тому, что в организме имеется 2 нормальные хромосомы номер 18 и еще часть генов с этих хромосом (сохранившийся фрагмент молекулы ДНК). В этом случае количество врожденных дефектов будет намного ниже. Наблюдается избыток не всей генетической информации, закодированной в 18-й хромосоме, а лишь ее части. Для пациентов с частичной трисомией 18 прогноз лучше, чем для детей с полной формой, но все равно остается неблагоприятным.
- Мозаичная форма. Мозаичная форма синдрома Эдвардса встречается в 5 – 7% случаев данного заболевания. Механизм ее появления отличается от других видов. Дело в том, что здесь дефект образовался уже после слияния сперматозоида и яйцеклетки. Обе гаметы (половые клетки) изначально имели нормальный кариотип и несли по одной хромосоме каждого вида. После слияния сформировалась клетка с нормальной формулой 46,ХХ или 46,XY. В процессе деления этой клетки произошел сбой. При удвоении генетического материала один из фрагментов получил дополнительную 18-ю хромосому. Таким образом, на определенном этапе сформировался зародыш, часть клеток которого имеют нормальный кариотип (например, 46,ХХ), а часть – кариотип синдрома Эдвардса (47,ХХ, 18+). Доля патологических клеток никогда не превышает 50%. Их число зависит от того, на каком этапе деления начальной клетки произошел сбой. Чем позже это происходит, тем меньше будет доля дефектных клеток. Форма получила название из-за того, что все клетки организма представляют собой своеобразную мозаику. Часть из них здорова, а часть – с тяжелой генетической патологией. Закономерности в распределении клеток в организме при этом не наблюдается, то есть все дефектные клетки не могут локализоваться только в одном месте, чтобы их можно было удалить. Общее состояние пациента при этом легче, чем при классической форме трисомии 18.
Согласно последним исследованиям хромосома номер 18 содержит 557 генов, которые кодируют не менее 289 различных белков. В процентном отношении это примерно 2,5% всего генетического материала. Нарушения, которые вызывает столь большой дисбаланс, очень серьезны. Неправильное количество белков предопределяет множество аномалий в развитии различных органов и тканей. В случае синдрома Эдвардса чаще других страдают кости черепа, некоторые отделы нервной системы, сердечно-сосудистая и мочеполовая система. По всей видимости, это связано с тем, что гены, расположенные на этой хромосоме, имеют отношение к развитию именно этих органов и систем.
Таким образом, основной и единственной причиной синдрома Эдвардса является наличие дополнительной молекулы ДНК. Наиболее часто (при классической форме болезни) она наследуется от одного из родителей. В норме каждая гамета (сперматозоид и яйцеклетка) содержат по 22 непарные соматические хромосомы, плюс одна половая. Женщина всегда передает ребенку стандартный набор 22+Х, а мужчина может передать 22+Х либо 22+Y. Это предопределяет пол ребенка. Половые клетки родителей образуются в результате разделения обычных клеток на два набора. В норме материнская клетка делится на две равные части, но иногда не все хромосомы делятся пополам. Если 18-я пара не разошлась по полюсам клетки, то одна из яйцеклеток (или одни из сперматозоидов) будет заранее дефектным. В нем будет не 23, а 24 хромосомы. В случае если именно эта клетка будет участвовать в оплодотворении, ребенок получит дополнительную 18-ю хромосому.
На неправильное деление клеток могут повлиять следующие факторы:
- Возраст родителей. Доказано, что вероятность хромосомных аномалий увеличивается прямо пропорционально с возрастом матери. При синдроме Эдвардса эта связь менее выражена, чем при других похожих патологиях (например, синдром Дауна). Но для женщин старше 40 лет риск родить ребенка с данной патологией в среднем в 6 – 7 раз выше. Подобная зависимость от возраста отца наблюдается в значительно меньшей степени.
- Курение и алкоголь. Такие вредные привычки как курение и злоупотребление алкоголем могут действовать на половую систему человека, влияя на деление половых клеток. Таким образом, регулярное употребление этих веществ (а также других наркотических препаратов) повышает риск неправильного распределения генетического материала.
- Прием лекарственных средств. Некоторые лекарственные препараты при неправильном приеме в первом триместре могут повлиять на деление зародышевых клеток и спровоцировать мозаичную форму синдрома Эдвардса.
- Заболевания половой сферы. Перенесенные инфекции с поражением репродуктивных органов могут отразиться на правильном делении клеток. Они повышают риск хромосомных и генетических заболеваний в целом, хотя специально для синдрома Эдвардса подобные исследования не проводились.
- Радиационное излучение. Облучение половых органов рентгеновским излучением или другими ионизирующими излучениями может повлечь генетические мутации. Особенно опасно такое внешнее воздействие в подростковом возрасте, когда деление клеток происходит наиболее активно. Частицы, формирующие излучение, легко проникают сквозь ткани и подвергают молекулу ДНК своеобразной «бомбардировке». Если это происходит в момент деления клетки, риск хромосомной мутации особенно высок.
Как выглядят новорожденные с синдромом Эдвардса?
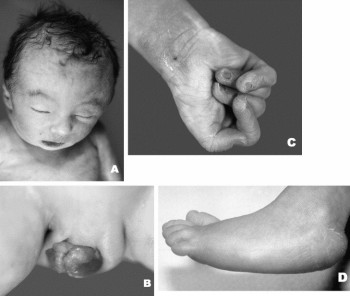 Как известно, диагностировать синдром Эдвардса можно до рождения, но в большинстве случаев данное заболевание обнаруживают непосредственно после рождения ребенка. У новорожденных с данной патологией имеется ряд ярко выраженных аномалий развития, которые иногда позволяют сразу заподозрить правильный диагноз. Подтверждение проводится позже при помощи специального генетического анализа.
Как известно, диагностировать синдром Эдвардса можно до рождения, но в большинстве случаев данное заболевание обнаруживают непосредственно после рождения ребенка. У новорожденных с данной патологией имеется ряд ярко выраженных аномалий развития, которые иногда позволяют сразу заподозрить правильный диагноз. Подтверждение проводится позже при помощи специального генетического анализа.Новорожденные с синдромом Эдвардса имеют следующие характерные аномалии развития:
- изменение формы черепа;
- изменение формы ушных раковин;
- аномалии развития неба;
- стопа-качалка;
- аномальная длина пальцев;
- изменение формы нижней челюсти;
- сращение пальцев;
- аномалии развития половых органов;
- флексорное положение кистей;
- дерматоглифические признаки.
Изменение формы черепа
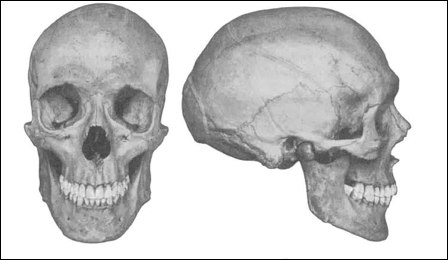 Типичным симптомом при синдроме Эдвардса является долихоцефалия. Так называется характерное изменение формы головы новорожденного ребенка, которое встречается и при некоторых других генетических заболеваниях. У долихоцефалов (детей с данным симптомом) более длинный и узкий череп. Точно подтверждается наличие этой аномалии с помощью специальных измерений. Определяют соотношение ширины черепа на уровне теменных костей к длине черепа (от выступа над переносицей до затылочного бугра). Если полученное соотношение меньше 75%, то данный ребенок относится к долихоцефалам. Сам по себе данный симптом не является серьезным нарушением. Это просто один из видов формы черепа, встречающийся и у абсолютно нормальных людей. Дети с синдромом Эдвардса в 80 – 85% случаев являются выраженными долихоцефалами, у которых диспропорцию длины и ширины черепа можно заметить и без специальных измерений.
Типичным симптомом при синдроме Эдвардса является долихоцефалия. Так называется характерное изменение формы головы новорожденного ребенка, которое встречается и при некоторых других генетических заболеваниях. У долихоцефалов (детей с данным симптомом) более длинный и узкий череп. Точно подтверждается наличие этой аномалии с помощью специальных измерений. Определяют соотношение ширины черепа на уровне теменных костей к длине черепа (от выступа над переносицей до затылочного бугра). Если полученное соотношение меньше 75%, то данный ребенок относится к долихоцефалам. Сам по себе данный симптом не является серьезным нарушением. Это просто один из видов формы черепа, встречающийся и у абсолютно нормальных людей. Дети с синдромом Эдвардса в 80 – 85% случаев являются выраженными долихоцефалами, у которых диспропорцию длины и ширины черепа можно заметить и без специальных измерений.Другим вариантом аномалии развития черепа является так называемая микроцефалия, при которой размеры головы в целом слишком малы по сравнению с остальным туловищем. Прежде всего, это касается не лицевого черепа (челюсти, скулы, глазницы), а именно черепной коробки, в которой располагается мозг. Микроцефалия менее характерна для синдрома Эдвардса, чем долихоцефалия, но она тоже встречается с более высокой частотой, чем среди здоровых людей.
Изменение формы ушной раковины
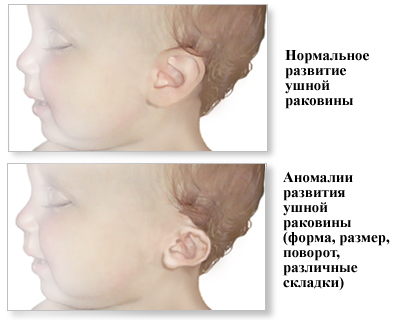 Если долихоцефалия может быть вариантом нормы, то патологии развития ушной раковины у детей с синдромом Эдвардса куда тяжелее. В определенной степени этот симптом наблюдается более чем у 95% детей с полной формой данного заболевания. При мозаичной форме его частота несколько меньше. Ушная раковина обычно располагается ниже, чем у нормальных людей (иногда ниже уровня глаз). Характерные выпуклости хряща, который формирует ушную раковину, плохо выражены или отсутствуют. Также могут отсутствовать мочка или козелок (небольшой выступающий участок хряща спереди от слухового отверстия). Сам слуховой проход обычно сужен, а примерно в 20 – 25% - вовсе отсутствует.
Если долихоцефалия может быть вариантом нормы, то патологии развития ушной раковины у детей с синдромом Эдвардса куда тяжелее. В определенной степени этот симптом наблюдается более чем у 95% детей с полной формой данного заболевания. При мозаичной форме его частота несколько меньше. Ушная раковина обычно располагается ниже, чем у нормальных людей (иногда ниже уровня глаз). Характерные выпуклости хряща, который формирует ушную раковину, плохо выражены или отсутствуют. Также могут отсутствовать мочка или козелок (небольшой выступающий участок хряща спереди от слухового отверстия). Сам слуховой проход обычно сужен, а примерно в 20 – 25% - вовсе отсутствует.Аномалии развития неба
 Небные отростки верхней челюсти в процессе развития эмбриона срастаются, формируя твердое небо. У детей с синдромом Эдвардса этот процесс нередко остается незавершенным. В том месте, где у нормальных людей располагается срединный шов (его можно прощупать посередине твердого неба языком) у них идет продольная щель.
Небные отростки верхней челюсти в процессе развития эмбриона срастаются, формируя твердое небо. У детей с синдромом Эдвардса этот процесс нередко остается незавершенным. В том месте, где у нормальных людей располагается срединный шов (его можно прощупать посередине твердого неба языком) у них идет продольная щель.Существует несколько вариантов данного дефекта:
- незаращение мягкого неба (задняя, глубокая часть неба, которая нависает над глоткой);
- частичное незаращение твердого неба (щель не тянется на протяжении всей верхней челюсти);
- полное незаращение твердого и мягкого неба;
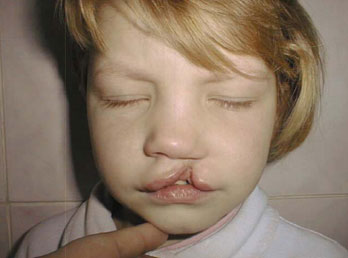
- полное незаращение неба и губы.
Данные дефекты развития известны также под названием волчья пасть, расщепление неба, заячья губа. Все они могут встречаться и не в рамках синдрома Эдвардса, однако у детей с этой патологией их частота особенно высока (почти 20% новорожденных). Значительно чаще (до 65% новорожденных) обладают другой особенностью, известной как высокое или готическое небо. Оно может быть отнесено к вариантам нормы, так как встречается и у здоровых людей.
Наличие расщепленного верхнего неба или верхней губы еще не подтверждает синдром Эдвардса. Этот порок развития может встречаться с довольно высокой частотой и самостоятельно без сопутствующих нарушений со стороны других органов и систем. Для исправления данной аномалии существует ряд стандартных хирургических вмешательств.
Стопа-качалка
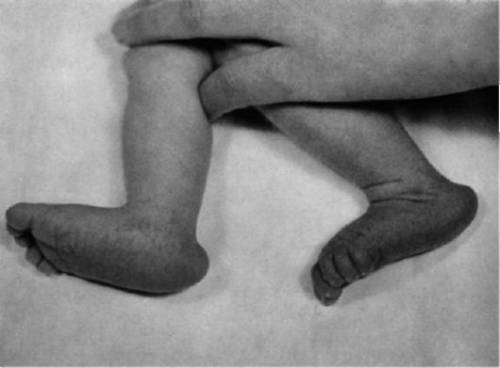 Так называется характерное изменение стопы, которое встречается, в основном, в рамках синдрома Эдвардса. Частота его при данном заболевании достигает 75%. Дефект заключается в неправильном взаимоположении таранной, пяточной и ладьевидной костей. Его относят к категории плоско-вальгусных деформаций стопы у детей.
Так называется характерное изменение стопы, которое встречается, в основном, в рамках синдрома Эдвардса. Частота его при данном заболевании достигает 75%. Дефект заключается в неправильном взаимоположении таранной, пяточной и ладьевидной костей. Его относят к категории плоско-вальгусных деформаций стопы у детей.Внешне стопа у новорожденного ребенка выглядит следующим образом. Пяточный бугор, на который опирается задняя часть стопы, выдается назад. Свод при этом может полностью отсутствовать. Это легко заметить, посмотрев на стопу с внутренней стороны. В норме там вырисовывается вогнутая линия, направляющаяся от пятки к основанию большого пальца. При стопе-качалке этой линии нет. Стопа плоская или даже выпуклая. Это и придает ей сходство с ножками кресла-качалки.
Аномальная длина пальцев
 У детей с синдромом Эдвардса на фоне изменений в строении стопы может наблюдаться ненормальная пропорция в длине пальцев ног. В частности, речь идет о большом пальце, который в норме является самым длинным. У новорожденных с данным синдромом он уступает по длине второму пальцу. Данный дефект можно заметить лишь при распрямлении пальцев и тщательном их рассмотрении. С возрастом, по мере роста ребенка, он становится более заметным. Поскольку укорочение большого пальца стопы встречается в основном при стопе-качалке, распространенность этих симптомов у новорожденных примерно одинакова.
У детей с синдромом Эдвардса на фоне изменений в строении стопы может наблюдаться ненормальная пропорция в длине пальцев ног. В частности, речь идет о большом пальце, который в норме является самым длинным. У новорожденных с данным синдромом он уступает по длине второму пальцу. Данный дефект можно заметить лишь при распрямлении пальцев и тщательном их рассмотрении. С возрастом, по мере роста ребенка, он становится более заметным. Поскольку укорочение большого пальца стопы встречается в основном при стопе-качалке, распространенность этих симптомов у новорожденных примерно одинакова.У взрослых укорочение большого пальца на ноге не имеет такой диагностической ценности. Подобный дефект может быть индивидуальной особенностью у здорового человека или следствием воздействия других факторов (деформация суставов, заболевания костей, ношение обуви, не соответствующей по размеру). В связи с этим данный признак нужно рассматривать как возможный симптом только у новорожденных детей при наличии других аномалий развития.
Изменение формы нижней челюсти
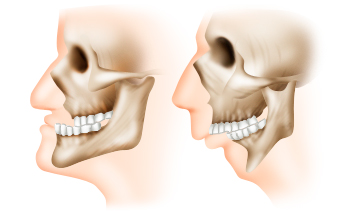 Изменения формы нижней челюсти у новорожденных встречаются почти в 70% случаев. В норме подбородок у детей не выступает вперед так, как у взрослых, но у больных с синдромом Эдвардса он слишком уж сильно втянут. Это происходит из-за недоразвития нижней челюсти, которое носит название микрогнатия (микрогения). Данный симптом встречается и при других врожденных заболеваниях. Не так уж редко можно встретить и взрослых людей с похожими чертами лица. При отсутствии сопутствующих патологий это считается вариантом нормы, хоть и ведет к некоторым трудностям.
Изменения формы нижней челюсти у новорожденных встречаются почти в 70% случаев. В норме подбородок у детей не выступает вперед так, как у взрослых, но у больных с синдромом Эдвардса он слишком уж сильно втянут. Это происходит из-за недоразвития нижней челюсти, которое носит название микрогнатия (микрогения). Данный симптом встречается и при других врожденных заболеваниях. Не так уж редко можно встретить и взрослых людей с похожими чертами лица. При отсутствии сопутствующих патологий это считается вариантом нормы, хоть и ведет к некоторым трудностям.У новорожденных с микрогнатией обычно быстро появляются следующие проблемы:
- невозможность долго держать рот закрытым (подтекание слюны);
- затруднения при кормлении;
- позднее развитие зубов и неправильное их расположение.
Сращение пальцев
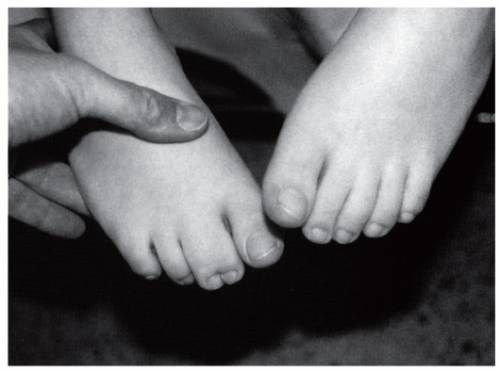 Сращение пальцев, или по-научному синдактилия, наблюдается приблизительно у 45% новорожденных. Чаще всего эта аномалия затрагивает пальцы ног, но встречается и синдактилия на руках. В легких случаях сращение образовано кожной складкой наподобие короткой перепонки. В более тяжелых случаях наблюдается сращение мостиками костной ткани.
Сращение пальцев, или по-научному синдактилия, наблюдается приблизительно у 45% новорожденных. Чаще всего эта аномалия затрагивает пальцы ног, но встречается и синдактилия на руках. В легких случаях сращение образовано кожной складкой наподобие короткой перепонки. В более тяжелых случаях наблюдается сращение мостиками костной ткани.Синдактилия встречается не только при синдроме Эдвардса, но и при многих других хромосомных заболеваниях. Известны и случаи, когда этот порок развития являлся единственным, и в остальном больной ничем не отличался от нормальных детей. В связи с этим сращение пальцев является лишь одним из возможных признаков синдрома Эдвардса, который помогает заподозрить диагноз, но не подтверждает его.
Аномалии развития половых органов
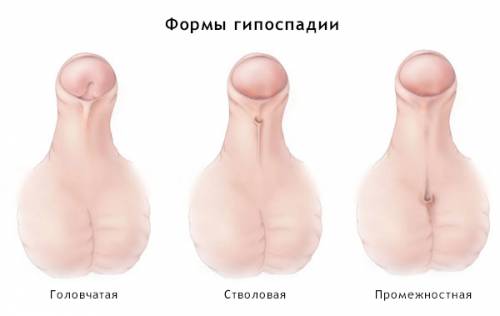 Непосредственно после родов у новорожденных с синдромом Эдвардса иногда можно наблюдать аномалии развития внешних половых органов. Как правило, они сочетаются с дефектами развития всего мочеполового аппарата, однако без специальных диагностических мероприятий это установить невозможно. Наиболее же частыми аномалиями, заметными внешне, являются недоразвитие полового члена у мальчиков и гипертрофия (увеличение в размерах) клитора у девочек. Они встречаются примерно в 15 – 20% случаев. Несколько реже может наблюдаться аномальное расположение мочеиспускательного канала (гипоспадия) или отсутствие яичек в мошонке у мальчиков (крипторхизм).
Непосредственно после родов у новорожденных с синдромом Эдвардса иногда можно наблюдать аномалии развития внешних половых органов. Как правило, они сочетаются с дефектами развития всего мочеполового аппарата, однако без специальных диагностических мероприятий это установить невозможно. Наиболее же частыми аномалиями, заметными внешне, являются недоразвитие полового члена у мальчиков и гипертрофия (увеличение в размерах) клитора у девочек. Они встречаются примерно в 15 – 20% случаев. Несколько реже может наблюдаться аномальное расположение мочеиспускательного канала (гипоспадия) или отсутствие яичек в мошонке у мальчиков (крипторхизм).Флексорное положение кистей
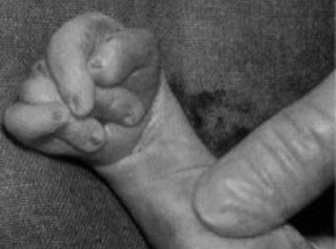 Флексорное положение кистей – это особое расположение пальцев, вызванное не столько структурными нарушениями в области кисти, сколько повышенным тонусом мышц. Сгибатели пальцев и кисти постоянно напряжены, из-за чего большой палец и мизинец как бы прикрывают остальные пальцы, которые при этом прижаты к ладони. Данный симптом наблюдается при многих врожденных патологиях и не является характерным именно для синдрома Эдвардса. Тем не менее, при обнаружении кисти подобной формы необходимо предполагать эту патологию. При ней флексорное положение пальцев наблюдается почти у 90% новорожденных.
Флексорное положение кистей – это особое расположение пальцев, вызванное не столько структурными нарушениями в области кисти, сколько повышенным тонусом мышц. Сгибатели пальцев и кисти постоянно напряжены, из-за чего большой палец и мизинец как бы прикрывают остальные пальцы, которые при этом прижаты к ладони. Данный симптом наблюдается при многих врожденных патологиях и не является характерным именно для синдрома Эдвардса. Тем не менее, при обнаружении кисти подобной формы необходимо предполагать эту патологию. При ней флексорное положение пальцев наблюдается почти у 90% новорожденных.Дерматоглифические признаки
 При многих хромосомных аномалиях у новорожденных имеются характерные дерматоглифические изменения (аномальные узоры и складки на коже ладоней). При синдроме Эдвардса некоторые признаки можно обнаружить почти в 60% случаев. Они имеют значение в основном для предварительной диагностики при мозаичной или частичной форме болезни. При полной трисомии 18 к дерматоглифике не прибегают, так как для подозрения синдрома Эдвардса хватает других, более заметных аномалий развития.
При многих хромосомных аномалиях у новорожденных имеются характерные дерматоглифические изменения (аномальные узоры и складки на коже ладоней). При синдроме Эдвардса некоторые признаки можно обнаружить почти в 60% случаев. Они имеют значение в основном для предварительной диагностики при мозаичной или частичной форме болезни. При полной трисомии 18 к дерматоглифике не прибегают, так как для подозрения синдрома Эдвардса хватает других, более заметных аномалий развития.Основными дерматоглифическими признаками синдрома Эдвардса являются:
- дуги на подушечках пальцев располагаются с большей частотой, нежели у здоровых людей;
- кожная складка между последней (ногтевой) и предпоследней (срединной) фалангами пальцев отсутствует;
- у 30% новорожденных на ладони имеется так называемая поперечная борозда (обезьянья линия, линия Симиан).
Помимо вышеперечисленных признаков существует еще целый ряд возможных аномалий развития, которые могут помочь в предварительной диагностике синдрома Эдвардса. По некоторым данными при подробном внешнем осмотре можно обнаружить до 50 внешних признаков. Сочетание наиболее частых симптомов, представленных выше, с высокой вероятностью говорит о наличии у ребенка этой тяжелой патологии. При мозаичном варианте синдрома Эдвардса множественных аномалий может и не быть, однако наличие даже одного из них является показанием к проведению специального генетического теста.
Подпишитесь на Здоровьесберегающий видеоканал


Как выглядят дети с синдромом Эдвардса?
 У детей с синдромом Эдвардса по мере взросления обычно обнаруживаются самые разные сопутствующие патологии. Их симптомы начинают проявляться уже через несколько недель после рождения. Эти симптомы могут оказаться первым проявлением синдрома, так как при мозаичном варианте в редких случаях болезнь может остаться незамеченной непосредственно после рождения. Тогда диагностика заболевания усложняется.
У детей с синдромом Эдвардса по мере взросления обычно обнаруживаются самые разные сопутствующие патологии. Их симптомы начинают проявляться уже через несколько недель после рождения. Эти симптомы могут оказаться первым проявлением синдрома, так как при мозаичном варианте в редких случаях болезнь может остаться незамеченной непосредственно после рождения. Тогда диагностика заболевания усложняется.Большинство внешних проявлений синдрома, замеченных при рождении, остаются и становятся более заметными. Речь идет о форме черепа, стопе-качалке, деформации ушной раковины и т. п. Постепенно к ним начинают прибавляться и другие внешние проявления, которые невозможно было заметить сразу после рождения. В данном случае речь идет о признаках, которые могут появиться у детей в первый год жизни.
Дети с синдромом Эдвардса имеют следующие внешние особенности:
- отставание в физическом развитии;
- косолапость;
- аномальный тонус мышц;
- ненормальные эмоциональные реакции.
Отставание в физическом развитии
Отставание в физическом развитии объясняется низкой массой тела ребенка при рождении (всего 2000 – 2200 г при нормальном сроке беременности). Значительную роль играет и генетический дефект, который не позволяет всем системам организма нормально и гармонично развиваться. Основные показатели, по которым оценивают рост и развитие ребенка, сильно снижены.Заметить отставание ребенка можно по следующим антропометрическим показателям:
- рост ребенка;
- вес ребенка;
- окружность грудной клетки;
- окружность головы (данный показатель может быть в норме или даже увеличен, но на него нельзя полагаться из-за врожденной деформации черепа).
Косолапость
Косолапость является следствием деформации костей и суставов стоп, а также отсутствия нормального контроля со стороны нервной системы. Дети с трудом начинают ходить (большинство не доживает до этого этапа из-за врожденных пороков развития). Внешне о наличии косолапости можно судить по деформации стоп, ненормальному положению ног в состоянии покоя.Аномальный тонус мышц
Аномальный тонус, который при рождении вызывает флексорное положение кисти, по мере роста начинает проявляться и на других группах мышц. Чаще всего у детей с синдромом Эдвардса сила мышц снижена, они вялые и лишены нормального тонуса. В зависимости от характера повреждений центральной нервной системы некоторые группы могут иметь повышенный тонус, что проявляется спастическими сокращениями этих мышц (например, сгибатели рук или разгибатели ног). Внешне это проявляется отсутствием минимальной координации движений. Иногда спастические сокращения ведут к ненормальным перегибам конечностей или даже к вывихам.Ненормальные эмоциональные реакции
Отсутствие или ненормальное проявление каких-либо эмоций является следствием аномалий в развитии некоторых отделов мозга (чаще всего мозжечка и мозолистого тела). Эти изменения приводят к серьезному отставанию в умственном развитии, которое наблюдается у всех без исключения детей с синдромом Эдвардса. Внешне низкий уровень развития проявляется характерным «отсутствующим» выражением лица, отсутствием эмоционального ответа на внешние раздражители. Ребенок плохо поддерживает визуальный контакт (не следит за движущимся перед глазами пальцем и т. п.). Отсутствие реакции на резкие звуки может быть следствием поражения как нервной системы, так и слухового аппарата. Все эти признаки обнаруживаются по мере роста ребенка в первые месяцы жизни.Как выглядят взрослые с синдромом Эдвардса?
 В подавляющем большинстве случаев дети, рожденные с синдромом Эдвардса, не доживают до взрослого возраста. При полной форме этого заболевания, когда лишняя хромосома присутствует в каждой клетке тела, 90% детей умирает в возрасте до 1 года из-за серьезных аномалий развития внутренних органов. Даже при условии хирургического исправления возможных дефектов и качественном уходе их организм более подвержен инфекционным заболеваниям. Этому способствуют и нарушения питания, которые встречаются у большинства детей. Все это объясняет высочайшую смертность при синдроме Эдвардса.
В подавляющем большинстве случаев дети, рожденные с синдромом Эдвардса, не доживают до взрослого возраста. При полной форме этого заболевания, когда лишняя хромосома присутствует в каждой клетке тела, 90% детей умирает в возрасте до 1 года из-за серьезных аномалий развития внутренних органов. Даже при условии хирургического исправления возможных дефектов и качественном уходе их организм более подвержен инфекционным заболеваниям. Этому способствуют и нарушения питания, которые встречаются у большинства детей. Все это объясняет высочайшую смертность при синдроме Эдвардса.При более легкой мозаичной форме, когда лишь часть клеток в организме содержит аномальный набор хромосом, выживаемость несколько больше. Однако даже в этих случаях до взрослого возраста доживают единичные пациенты. Их внешний вид определяется врожденными аномалиями, которые присутствовали при рождении (заячья губа, деформированная ушная раковина, и др.). Основным же симптомом, присутствующим у всех без исключения детей, является серьезнейшее отставание в умственном развитии. Дожив до взрослого возраста, ребенок с синдромом Эдвардса является глубоким олигофреном (IQ менее 20, что соответствует самой тяжелой степени умственной отсталости). В целом же в медицинской литературе описываются единичные случаи, когда дети с синдромом Эдвардса доживали до совершеннолетнего возраста. Из-за этого накоплено слишком мало объективных данных, чтобы говорить о внешних признаках этого заболевания у взрослых.
Диагностика генетической патологии
 В настоящее время существуют три основных этапа диагностики синдрома Эдвардса, каждый из которых включает несколько возможных методов. Поскольку данное заболевание является неизлечимым, родителям следует обратить внимание на возможности этих методов и воспользоваться ими. Большинство анализов проводится в специальных центрах пренатальной диагностики, где имеется вся необходимая техника для поиска генетических заболеваний. Однако даже консультация у врача-генетика или неонатолога может оказаться полезной.
В настоящее время существуют три основных этапа диагностики синдрома Эдвардса, каждый из которых включает несколько возможных методов. Поскольку данное заболевание является неизлечимым, родителям следует обратить внимание на возможности этих методов и воспользоваться ими. Большинство анализов проводится в специальных центрах пренатальной диагностики, где имеется вся необходимая техника для поиска генетических заболеваний. Однако даже консультация у врача-генетика или неонатолога может оказаться полезной.Диагностика синдрома Эдвардса возможна на следующих этапах:
- диагностика до момента зачатия;
- диагностика во время внутриутробного развития;
- диагностика после рождения.
Диагностика до момента зачатия
Диагностика до момента зачатия ребенка является идеальным вариантом, но, к сожалению, на современном этапе развития медицины ее возможности очень ограничены. Врачи могут с помощью нескольких методов предположить повышенную вероятность рождения ребенка с хромосомным заболеванием, но не более того. Дело в том, что при синдроме Эдвардса, в принципе, нарушения у родителей обнаружить нельзя. Дефектная половая клетка с 24 хромосомами является лишь одной из многих тысяч. Поэтому сказать наверняка до момента зачатия, родится ли ребенок с данным заболеванием, нельзя.Основными методами диагностики до момента зачатия являются:
- Семейный анамнез. Семейный анамнез представляет собой подробный опрос обоих родителей об их родословной. Врача интересуют любые случаи наследственных (и особенно хромосомных) заболеваний в семье. Если хотя бы один из родителей припоминает случай трисомии (синдром Эдвардса, Дауна, Патау), это сильно повышает вероятность рождения больного ребенка. Однако риск все равно составляет не более 1%. При повторных случаях этих заболеваний у предков риск многократно возрастает. По сути, анализ сводится к консультации у неонатолога или генетика. Предварительно родители могут постараться собрать более подробную информацию о своих предках (желательно на 3 – 4 колена). Это повысит точность данного метода.
- Обнаружение факторов риска. Основным фактором риска, который объективно повышает риск хромосомных аномалий, является возраст матери. Как уже говорилось выше, у матерей после 40 лет вероятность рождения ребенка с синдромом Эдвардса многократно возрастает. По некоторым данным, после 45 лет (возраст матери) почти каждая пятая беременность сопровождается хромосомной патологией. Большинство из них заканчивается выкидышем. Другими факторами являются перенесенные инфекционные заболевания, хронические болезни, вредные привычки. Однако их роль в диагностике значительно более низкая. Точного ответа на вопрос, будет ли зачат ребенок с синдромом Эдвардса, этот метод тоже не дает.
- Генетический анализ родителей. Если предыдущие методы сводились к опросу родителей, то генетический анализ представляет собой полноценное исследование, которое требует наличия специальной аппаратуры, реактивов и квалифицированных специалистов. У родителей берется кровь, из которой в лаборатории выделяют лейкоциты. После обработки специальными веществами в этих клетках становятся хорошо видны хромосомы на стадии деления. Таким образом составляется кариотип родителей. В подавляющем большинстве случаев он нормальный (при хромосомных нарушениях, которые могут быть здесь обнаружены, вероятность продолжения рода ничтожно мала). Кроме того с помощью специальных маркеров (фрагменты молекулярных цепочек) можно обнаружить участки ДНК с дефектными генами. Однако здесь будут обнаружены не хромосомные нарушения, а генетические мутации, которые не влияют напрямую на вероятность синдрома Эдвардса. Таким образом, генетический анализ родителей до момента зачатия, несмотря на сложность и высокую стоимость, также не дает однозначного ответа относительно прогнозов на данную патологию.
Диагностика во время внутриутробного развития
В период внутриутробного развития существует несколько способов, которые могут прямо или косвенно подтвердить наличие у зародыша хромосомной патологии. Точность этих методов значительно выше, так как врачи имеют дело не с родителями, а с самим организмом плода. Для изучения доступен как сам зародыш, так и его клетки с собственным ДНК. Данный этап называется также пренатальной диагностикой и является наиболее важным. В это время можно подтвердить диагноз, предупредить родителей о наличии патологии и при необходимости прервать беременность. Если же женщина решит рожать и новорожденный будет жив, то врачи получат возможность заранее подготовиться к оказанию ему необходимой помощи.Основными методами исследования в рамках пренатальной диагностики являются:
- Ультразвуковое исследование (УЗИ). Данный метод является неинвазивным, то есть не предполагает повреждения тканей матери или плода. Он полностью безопасен и рекомендован для всех беременных женщин в рамках пренатальной диагностики (независимо от их возраста или повышенного риска для хромосомных заболеваний). Стандартная программа предполагает, что УЗИ надо делать трижды (на 10 – 14, 20 – 24 и 32 – 34 неделе беременности). Если лечащий врач предполагает возможность врожденных аномалий развития, могут быть проведены и незапланированные УЗИ. О синдроме Эдвардса может говорить отставание плода в размерах и массе, большое количество околоплодных вод, видимые аномалии развития (микроцефалия, деформация костей). Эти нарушения с высокой вероятностью говорят о тяжелых генетических заболеваниях, но синдром Эдвардса окончательно подтвердить все же невозможно.
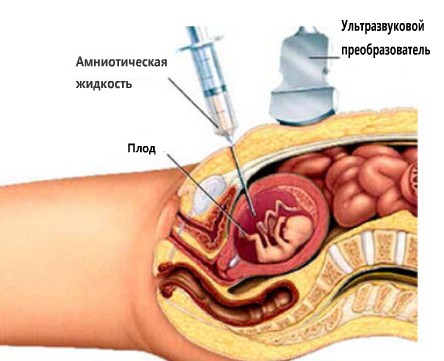
- Амниоцентез. Амниоцентез представляет собой цитологический (клеточный) анализ околоплодных вод. Врач аккуратно вводит специальную иглу под контролем аппарата УЗИ. Прокол делается в месте, где нет петель пупочного канатика. С помощью шприца берется необходимое для исследования количество амниотической жидкости. Процедуру можно проводить во всех триместрах беременности, но оптимальным сроком для диагностики хромосомных нарушений является период после 15 недели беременности. Частота осложнений (вплоть до спонтанного прерывания беременности) составляет до 1%, поэтому процедуру не стоит проводить при отсутствии каких-либо показаний. После забора околоплодных вод проводится обработка полученного материала. В них жидкости имеются клетки с поверхности кожи малыша, которые содержат образцы его ДНК. Именно их и проверяют на наличие генетических заболеваний.
- Кордоцентез. Кордоцентез представляет собой наиболее информативный метод пренатальной диагностики. После обезболивания и под контролем аппарата УЗИ врач прокалывает с помощью специальной иглы сосуд, проходящий в пуповине. Таким образом, получают образец крови (до 5 мл) развивающегося ребенка. Техника выполнения анализа аналогична таковой для взрослых. Данный материал можно с высокой точностью исследовать на предмет различных генетических аномалий. В том числе можно сделать кариотипирование плода. При наличии дополнительной 18-й хромосомы можно говорить о подтвержденном синдроме Эдвардса. Данный анализ рекомендуется проводить после 18-й недели беременности (оптимально 22 – 25 недели). Частота возможных осложнений после кордоцентеза составляет 1,5 – 2%.
- Биопсия хориона. Хорион представляет собой одну из зародышевых оболочек, содержащую клетки с генетической информацией плода. Данное исследование предполагает пункцию матки под наркозом через переднюю брюшную стенку. С помощью специальных биопсийных щипцов берут образец ткани для анализа. Затем проводится стандартное генетическое исследование полученного материала. Для диагностики синдрома Эдвардса делается кариотипирование. Оптимальным сроком для проведения биопсии хориона считают 9 – 12 неделю беременности. Частота осложнений составляет 2 – 3%. Основным преимуществом, отличающим его от других методов, является скорость получения результата (уже через 2 – 4 дня).
Диагностика после рождения
Диагностика синдрома Эдвардса после рождения является наиболее легкой, быстрой и точной. К сожалению, на этот момент уже произошло появление на свет ребенка с тяжелой генетической патологией, эффективного лечения которой в наше время пока не существует. Если на этапе пренатальной диагностики заболевание не было выявлено (либо соответствующие исследования не проводились), то подозрение на синдром Эдвардса появляется сразу после рождения. Ребенок обычно доношен или даже переношен, но его масса все равно ниже среднего показателя. Кроме того, обращают на себя внимание некоторые врожденные дефекты, о которых говорилось выше. Если их замечают, проводят генетический анализ для подтверждения диагноза. У ребенка берется кровь для анализа. Однако на данном этапе подтвердить наличие синдрома Эдвардса – неглавная проблема.Основной задачей при рождении ребенка с этой патологией является обнаружение аномалий в развитии внутренних органов, которые обычно приводят к смерти в первые месяцы жизни. Именно на их поиск направлено большинство диагностических процедур непосредственно после рождения.
Для обнаружения пороков в развитии внутренних органов применяют следующие методы исследования:
- ультразвуковое исследование брюшной полости;
- эхокардиография;
- общий анализ крови и биохимический анализ крови;
- общий анализ мочи;
- компьютерная томография или магнитно-резонансная томография;
- рентгенография.
Таким образом, можно заключить, что существует много методов диагностики синдрома Эдвардса. Некоторые из них (амниоцентез, кордоцентез и др.) представляют определенную опасность осложнений и не проводятся без специальных показаний. Основными показаниями считается наличие в роду случаев хромосомных заболеваний и возраст матери старше 35 лет. Программа диагностики и ведения пациентки на всех этапах беременности может быть изменена лечащим врачом по необходимости.
Прогноз для детей с синдромом Эдвардса
 Учитывая множественные нарушения развития, которые присущи синдрому Эдвардса, прогноз для новорожденных с этим диагнозом почти всегда неблагоприятный. Статистические данные (из различных независимых исследований) говорят, что больше половины детей (50 – 55%) не доживают до трехмесячного возраста. Первый день рождения удается отпраздновать меньше чем десяти процентам малышей. Те дети, которые доживают до старшего возраста, имеют серьезнейшие проблемы со здоровьем и нуждаются в постоянном уходе. Для продления жизни нередко необходимы сложные хирургические операции на сердце, почках или других внутренних органах. Исправление врожденных дефектов и постоянный квалифицированный уход, по сути, являются единственным лечением. У детей с классической формой синдрома Эдвардса (полной трисомией 18) шансов на нормальное детство или сколько-нибудь длительную жизнь практически нет.
Учитывая множественные нарушения развития, которые присущи синдрому Эдвардса, прогноз для новорожденных с этим диагнозом почти всегда неблагоприятный. Статистические данные (из различных независимых исследований) говорят, что больше половины детей (50 – 55%) не доживают до трехмесячного возраста. Первый день рождения удается отпраздновать меньше чем десяти процентам малышей. Те дети, которые доживают до старшего возраста, имеют серьезнейшие проблемы со здоровьем и нуждаются в постоянном уходе. Для продления жизни нередко необходимы сложные хирургические операции на сердце, почках или других внутренних органах. Исправление врожденных дефектов и постоянный квалифицированный уход, по сути, являются единственным лечением. У детей с классической формой синдрома Эдвардса (полной трисомией 18) шансов на нормальное детство или сколько-нибудь длительную жизнь практически нет.При частичной трисомии или мозаичной форме синдрома прогноз несколько лучше. Средняя продолжительность жизни при этом увеличивается до нескольких лет. Это объясняется тем, то аномалии развития при более легких формах не ведут так быстро к смерти ребенка. Тем не менее, основная проблема, а именно серьезное отставание в умственном развитии, присуща всем без исключения больным. При достижении подросткового возраста нет шансов ни на продолжение потомства (половая зрелость обычно не наступает), ни на возможность работы (даже механической, не требующей особых навыков). Существуют специальные центры для ухода за детьми с врожденными заболеваниями, где больным с синдромом Эдвардса обеспечивают уход и по возможности способствуют их интеллектуальному развитию. При достаточных усилиях со стороны врачей и родителей ребенок, проживший больше года, может научиться улыбаться, реагировать на движение, самостоятельно поддерживать положение тела или питаться (при отсутствии пороков системы пищеварения). Таким образом, признаки развития все же наблюдаются.
Высокая детская смертность при данном заболевании объясняется большим количеством пороков развития внутренних органов. Они незаметны непосредственно при рождении, но присутствуют практически у всех больных. В первые месяцы жизни дети обычно умирают от остановки сердца или дыхания.
Чаще всего пороки развития наблюдаются в следующих органах и системах:
- опорно-двигательный аппарат (кости и суставы, включая череп);
- сердечно-сосудистая система;
- центральная нервная система;
- пищеварительная система;
- мочеполовая система;
- другие нарушения.
Опорно-двигательный аппарат
Основными пороками в развитии опорно-двигательного аппарата является аномальное положение пальцев и искривление стоп. В бедренном суставе наблюдается сведение ног таким образом, что колени почти соприкасаются, а стопы смотрят немного в стороны. Нередко у детей с синдромом Эдвардса обнаруживается необычно короткая грудина. Это деформирует грудную клетку в целом и создает проблемы с дыханием, которые усугубляются по мере роста, даже если сами легкие не затронуты.Дефекты развития черепа являются в основном косметическими. Однако такие пороки как волчья пасть, заячья губа и высокое небо создают серьезные трудности с кормлением ребенка. Нередко до проведения операций по исправлению этих дефектов ребенка переводят на парентеральное питание (в виде капельниц с питательными растворами). Другим вариантом является использование гастростомы – специального зонда, через который пища попадает прямо в желудок. Его установление требует отдельного хирургического вмешательства.
В целом пороки развития опорно-двигательного аппарата не создают прямой угрозы для жизни ребенка. Однако косвенно они влияют на его рост и развитие. Частота таких изменений у больных синдромом Эдвардса составляет около 98%.
Сердечно-сосудистая система
Пороки развития сердечно-сосудистой системы являются основной причиной смерти в раннем детском возрасте. Дело в том, что подобные нарушения встречаются почти в 90% случаев. Чаще всего они серьезно нарушают процесс транспорта крови по организму, приводя к выраженной сердечной недостаточности. Большинство сердечных патологий может быть исправлено хирургическим путем, но не каждому ребенку можно провести такую сложную операцию.Наиболее частыми аномалиями со стороны сердечно-сосудистой системы являются:
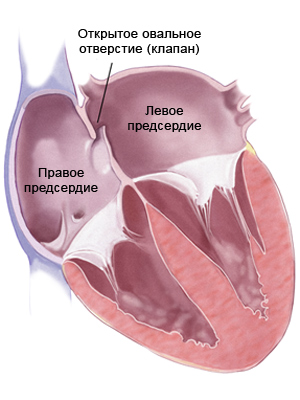
- незаращение межпредсердной перегородки;
- незаращение межжелудочковой перегородки;
- сращение створок клапанов (или, наоборот, их недоразвитие);
- коарктация (сужение) аорты.
Центральная нервная система
Самым характерным пороком со стороны центральной нервной системы является недоразвитие мозолистого тела и мозжечка. Это причина самых разнообразных нарушений, в том числе и умственной отсталости, которая наблюдается у 100% детей. Кроме того, нарушения на уровне головного и спинного мозга вызывают аномальный тонус мышц и предрасположенность к судорогам или спастическим сокращениям мышц.Пищеварительная система
Частота пороков пищеварительной системы при синдроме Эдвардса составляет до 55%. Чаще всего эти аномалии развития представляют серьезную угрозу жизни ребенка, поскольку не позволяют ему нормально усваивать питательные вещества. Питание же в обход естественных органов пищеварения сильно ослабляет организм и усугубляет состояние ребенка.Наиболее частыми пороками развития со стороны пищеварительной системы являются:
- дивертикул Меккеля (слепой отросток в тонкой кишке);
- атрезия пищевода (зарастание его просвета, из-за чего пища не проходит в желудок);
- атрезия желчных путей (накопление желчи в пузыре).
Мочеполовая система
Наиболее серьезные пороки со стороны мочеполовой системы связаны с нарушением работы почек. В ряде случаев наблюдается атрезия мочеточников. Почка с одной стороны может быть дублирована или сращена с лежащими рядом тканями. Если имеет место нарушение фильтрации, в организме со временем начинают накапливаться токсичные продукты жизнедеятельности. Кроме того может наблюдаться рост артериального давления и нарушения в работе сердца. Серьезные аномалии развития почек представляют прямую угрозу для жизни.Другие нарушения
Другими возможными нарушениями развития являются грыжи (пупочная, паховая). Могут обнаруживаться и дисковые грыжи позвоночника, которые приведут к неврологическим проблемам. Со стороны глаз иногда наблюдается микрофтальмия (маленький размер глазных яблок).Совокупность этих пороков развития предопределяет высокую детскую смертность. В большинстве случаев, если синдром Эдвардса диагностирован на ранних этапах беременности, врачи рекомендуют делать аборт по медицинским показаниям. Тем не менее, окончательное решение принимает сама пациентка. Несмотря на всю серьезность заболевания и плохой прогноз многие предпочитают надеяться на лучшее. Но, к сожалению, в ближайшее время серьезных сдвигов в методах диагностики и лечения синдрома Эдвардса, судя по всему, не предвидится.








Синдром Эдвардса (трисомия 18)
Неонатолог
Генетик
Синдром Шерешевского-Тёрнера
Синдром Дауна (трисомия 21)
Синдром кошачьего крика
Фенилкетонурия (ФКУ)
Муковисцидоз
Гипофизарный нанизм (карликовость)

Наши видеоканалы